
“Polycystic liver disease (PLD) is the result of embryonic ductal plate malformation of the intrahepatic biliary tree.The phenotype consists of numerous cysts spread throughout the liver parenchyma. Cystic bile duct malformations originating from the peripheral biliary tree are called Von Meyenburg complexes (VMC). In these patients embryonic remnants develop into small hepatic cysts and usually remain silent during life. Symptomatic PLD occurs mainly in
the context of isolated polycystic liver disease (PCLD) and autosomal dominant polycystic kidney disease (ADPKD).”
“Management of adult PLD is based on liver phenotype, severity of clinical features and quality of life. Conservative treatment is recommended for the majority of PLD patients. The primary aim is to halt cyst growth to allow abdominal decompression and ameliorate symptoms. Invasive procedures are required in a selective patient group with advanced PCLD, ADPKD or
liver failure. Pharmacological therapy by somatostatin analogues lead to beneficial outcome of PLD in terms of symptom relief and liver volume reduction.”
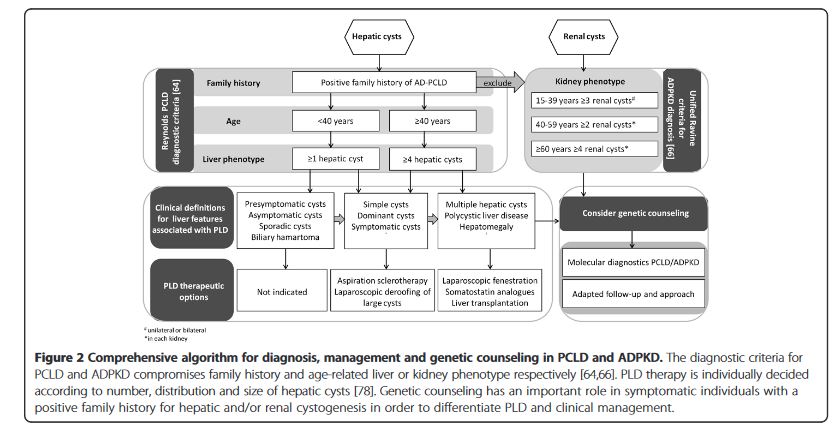
“PLD compromises a clinically heterogeneous liver phenotype identified in VMC, ADPKD and PCLD patients. Massively enlarged livers are present in a subset of ADPKD and PCLD. Genetics and environmental factors such as exogenous estrogen intake and number of
pregnancies contribute to disease progression. A considerable intra-familial variability in liver phenotype and extrahepatic features makes screening modalities uncertain in PCLD. Evaluation of PLD-related symptoms and quality of life are necessary to decide beneficial management.”
Cnossen, W.R., Drenth, J.P. Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J Rare Dis 9, 69 (2014).
